The thermodynamics of mixing
The dependence of the Gibbs energy of a mixture on its composition
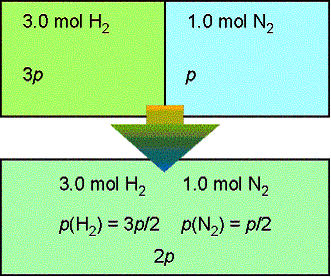
is given by ![]() . At constant T and p,
systems tend towards a lower Gibbs energy.
We will now quantitatively apply thermodynamics to the discussion of
spontaneous changes of composition, as in the mixing of two substances. (two gases mix spontaneously, so G must
decrease).
. At constant T and p,
systems tend towards a lower Gibbs energy.
We will now quantitatively apply thermodynamics to the discussion of
spontaneous changes of composition, as in the mixing of two substances. (two gases mix spontaneously, so G must
decrease).
The Gibbs energy of mixing
Two perfect gases in two identical containers with amounts
nA and nB, both at the
same T and p.

The difference Gf -Gi , the Gibbs
energy of mixing, DmixG, is
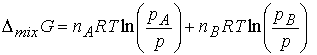
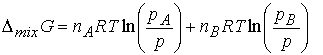
The chemical potential
terms cancel because they don’t change in this process.
Now we use Dalton’s law to write nJ = xJn and pJ/p = xJ, so
![]()
Because mole fractions are never greater than 1, the logarithms
are negative, and Gibbs energy of mixing < 0. Thus perfect gases mix spontaneously in all proportions.
Example
A container is divided into two equal compartments (figure
below). One contains 3.0 mol H2 at 25 0 C.; the other
contains 1.0 mol N2 at 25 0 C. Calculate the
Gibbs energy of mixing when the partition is removed. Assume perfect gas behavior.

Method: We first calculate
the initial Gibbs energy from
chemical potentials. We
need the pressure of each gas. Let p =
pressure of nitrogen, and the pressure of hydrogen can be found as a multiple
of p. p can be computed from the gas laws.
Next, calculate the Gibbs energy for the system when the partition is
removed. The volume of each gas doubles
so its partial pressure falls by a factor of two.
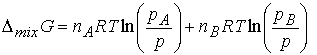
Answer: Use the expression for DmixG
Given the initial pressure of nitrogen is p, the pressure
of hydrogen is 3p; In the final
state the partial pressure of nitrogen is p/2 and hydrogen is 3p/2. Therefore
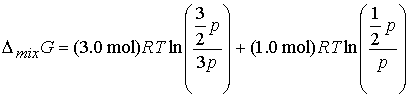
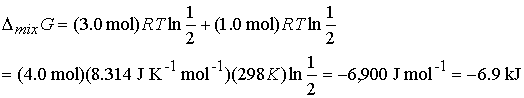
Other thermodynamic mixing functions
dG = -SdT + Vdp
Because ![]() it follows that DmixS is
it follows that DmixS is
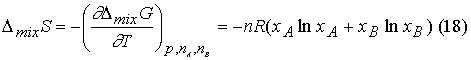
Because x <1,
ln(x) is < 0, and so DmixS > 0 for all
compositions. Mixing of perfect gases
is spontaneous.
The isothermal, isobaric enthalpy of mixing, DmixH, of two perfect
gases may be found from DG = DH - TDS.

DmixH of real gases or liq.-liq.,
solid-liq. mixtures are non-zero.
(eg. Ionic compounds in water)
7.3 The chemical potentials of liquids
We now investigate how the chemical potential of liquid mixtures
vary with composition. We use the fact
that at equilibrium the chemical potential of the liquid and vapor
components are equal.


Ideal solutions
Convention: quantities
relating to a pure substance will be denoted by a superscript *. No star – the mixture.
![]() , or
, or ![]() chemical potential
of pure A (liquid)
chemical potential
of pure A (liquid)
![]() chemical potential of
A in the standard state
chemical potential of
A in the standard state
![]() partial pressure of
A. We’ll keep pressure in bar so that
it can substitute for
partial pressure of
A. We’ll keep pressure in bar so that
it can substitute for 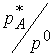 .
.
![]()
If another substance, a solute is present in the liquid,
the chemical potential of A in the liquid is mA and its vapor
pressure pA.
![]()
Next, we combine these two equations to estimate the standard chemical potential of the gas. Subtract the chemical potential of the vapor from the chemical potential of the liquid.
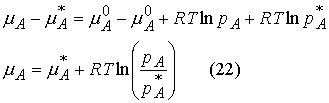
Raoult’s Law: For a liquid
mixture the ratio of the partial vapor pressure of each component to its vapor
pressure as a pure liquid, ![]()
![]() , is approximately equal to the mole fraction A in the liquid
mixture.
, is approximately equal to the mole fraction A in the liquid
mixture.
![]()
Mixtures that obey the law throughout are called ideal solutions.
For an ideal solution,
![]()

Solutions
will most likely behave ideally when they are chemical similar.
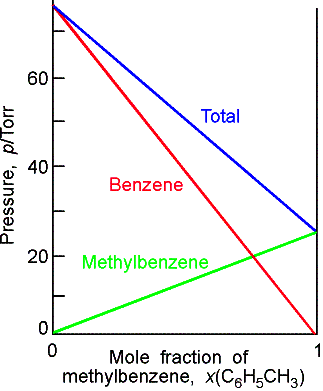


Molecular interpretation
Consider the rates at which molecules leave and return to a
liquid. Solute molecules at the air surface block some A molecules but in the
gas the A molecules are not inhibited from condensing because of the low
density of gases. The law reflects the fact that the presence of a second
component reduces the rate at which A molecules leave the surface of the
liquid but does not inhibit the rate at
which they return.
The rate at which A molecules
leave the surface is proportional to the concentration at the surface, which in
turn is proportional to xA:
rate of vaporization
= kxA
where k is a constant of proportionality. The rate at which molecules condense is
proportional to their concentration in the gas phase, which is proportional to
their partial pressure:
rate of condensation
= k’pA

At
equilibrium, the rates of vaporization and condensation are equal, so k’pA = kxA.
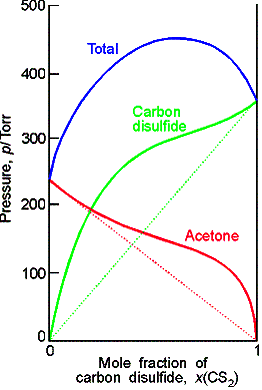
Ideal-dilute solutions
In ideal solutions the solute as well as the solvent obey Raoult’s law. For real solutions at low concentrations, although the vapor pressure of the solute is proportional to its mole fraction, the constant of proportionality is not the vapor pressure of the pure substance.
Henry’s law is: pB = xBKB
In this expression KB is an empirical constant (with
dimensions of pressure) chosen so that the plot of the vapor pressure of B
against its mole fraction is tangent to the experimental curve at xB = 0.
Ideal-dilute solutions are mixtures for which the solute obeys Henry’s
law and the solvent obeys Raoult’s law.
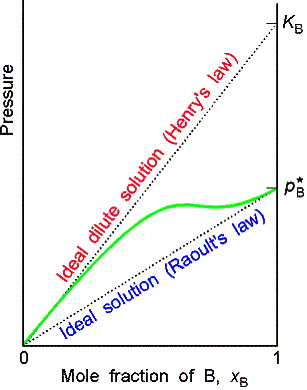

Molecular interpretation 7.2
The difference in behavior of the solute and the solvent at low
concentrations arises from the fact that in a dilute solution the solvent
molecules are in an environment very nearly like the pure liquid, but the
solute molecules are in an environment very different from the pure
solute.


Example
of a chloroform-acetone mixture.
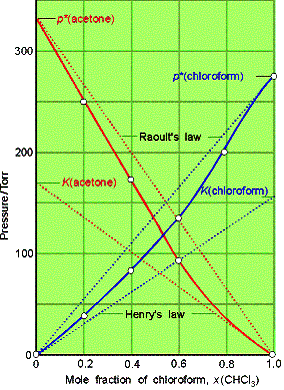


Example 7.4 Using Henry’s
Law
Estimate the molar solubility (the solubility in moles per liter)
of oxygen in water at 25 0C and a partial pressure of 160 Torr (its partial pressure
in the atmosphere at sea level).
Method The mole fraction
is given by Henry’s law as
xO = pO/K, where pO is the
partial pressure of oxygen. Use mole
fraction to solve for moles of O2.

Since the mole fraction of O2 is so low we can use the
moles of pure water for the moles of water in the solution.

Divers have to be concerned about the solubility of O2 and
N2 in blood since the concentration of dissolved gases in blood
increases as pressure is increased. If
a diver comes to the surface too quickly (where the solubility of N2
is lower) the N2 can form bubbles of gas in blood veins and capillaries
and cause the pain of the bends. Deep
sea divers breathe a mixture of 98% He, 2% O2. He has a much lower solubility in blood
than N2. At a pressure of 10
atm this gives an O2 partial pressure of about 0.2 atm (the pressure at atmosphere).